

作者:科学智能中心
编者按:AI 药物研发是人工智能未来应用的重要方向之一。自新冠病毒(SARS-CoV-2)首次爆发以来,新冠病毒的小分子药物研发备受关注,于近期举行的首届 AI 药物研发算法大赛便聚焦于此。在比赛中,来自微软研究院科学智能中心的团队,凭借创新的 AI 模型系统 AI2BMD 和 ViSNet 取得了绝佳的成绩,斩获桂冠。
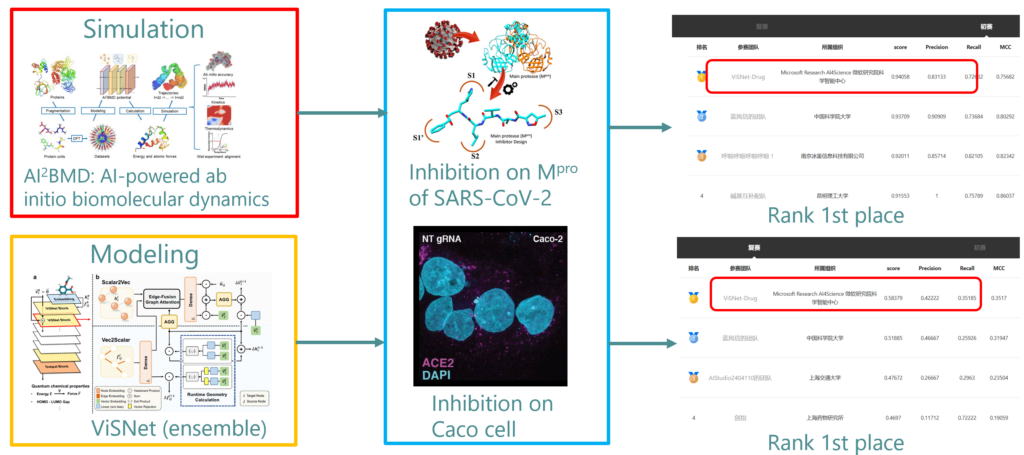
近日,由清华大学药学院、百度飞桨、百度智能云和临港实验室联袂主办的首届 AI 药物研发算法大赛公布了比赛结果,来自微软研究院科学智能中心的团队,利用研发的量子精度动力学模拟系统 AI2BMD 和通用分子三维结构网络 ViSNet 在初赛、复赛、决赛中均位列第一,并获得大赛的总冠军,展现了 AI 在促进药物研发方面的应用潜力。

本次大赛由中国药学会等业内权威机构鼎力支持,共有来自全球的878支团队参赛。作为一场全球性的技术创新活动,此次大赛聚焦于新冠病毒(SARS-CoV-2)小分子药物研发。事实上,自新冠病毒首次爆发以来,新冠病毒的小分子药物研发就备受关注。若要抵抗新冠病毒肆虐,深入了解病毒复制与感染机制至关重要。其中,新冠病毒主蛋白酶(Mpro)作为关键酶,负责感染过程中剪切病毒产生的蛋白质前体,促进病毒复制,所以主蛋白酶是一个潜在的治疗靶点,抑制其活性可有效干扰病毒的复制过程,为治疗方法提供突破口。因此,本次比赛的初赛阶段,参赛者需要使用深度学习、分子对接等方法进行建模,预测小分子抑制主蛋白酶活性的概率,复赛则重点关注小分子在 Caco 细胞上抑制新冠病毒复制的概率。
在初赛对新冠病毒主蛋白酶的药物预测中,面对常用分子对接软件无法有效区分正负样本与靶点蛋白结合自由能的问题,微软研究院科学智能中心团队利用了最新开发的 AI2BMD 模拟系统[1],将药物预测精度显著提升。AI2BMD 模拟系统实现了对超10000原子的各种蛋白质能能量和力的精确计算,并具有广泛的适用性。相较于密度泛函理论(DFT),AI2BMD 模拟系统的计算时间缩短了数个数量级。凭借几百纳秒的动力学模拟,AI2BMD 展现了在探索蛋白质构象空间、预测核磁共振实验数据以及模拟蛋白质折叠过程等方面的卓越能力。与传统分子对接、经典动力学模拟方法相比,AI2BMD 系统在计算结合自由能方面也有明显优势。
AI2BMD 模拟系统论文链接:
https://www.biorxiv.org/content/10.1101/2023.07.12.548519v1
复赛中,团队运用自主开发的分子建模几何深度学习模型 ViSNet [2] 化合物分子进行了表征学习。ViSNet 是 AI2BMD 模拟系统中的机器学习势能函数。作为一种等变的几何增强图神经网络,ViSNet 能在线性计算的复杂度下提取几何特征(距离、角度、二面角等)。在多个分子动力学基准(包括 MD17、rMD17 和 MD22)上,ViSNet 表现均优于其他先进方法,同时也在 QM9 和 Molecule3D 数据集上实现了卓越的量子化学性质预测。
分子建模几何深度学习模型 ViSNet 论文链接:
https://arxiv.org/abs/2210.16518v1
团队在复赛阶段,还利用自主研发的首个蛋白大分子全构象空间数据集 AIMD-Chig [3] 和小分子公开数据集 OGB 分别对蛋白和小分子的三维结构表征进行了预训练,然后通过多任务学习对模型进行微调。该方法不仅取得了最佳的预测精度,而且以大比分领先比赛的第二名团队。在最终的决赛答辩中,微软研究院科学智能中心团队的新冠药物预测算法方案取得了总分99.60分的绝佳成绩,相较比赛亚军90.76分、季军85.31分的最终成绩具有显著优势。
蛋白大分子全构象空间数据集 AIMD-Chig 论文链接:
https://www.nature.com/articles/s41597-023-02465-9
通过此次药物研发大赛,微软研究院科学智能中心开发的量子精度动力学模拟系统 AI2BMD 展现了出色的实际应用潜力。未来,AI2BMD 有望在生命活动的分子机理解释、药物设计、酶催化等方面进行更广泛的探索,助力 AI 药物研发的加速发展。
[1] Wang T, He X, Li M, et al. AI2BMD: efficient characterization of protein dynamics with ab initio accuracy. bioRxiv, 2023: 2023.07. 12.548519.
[2] Wang Y, Li S, Wang T, et al. ViSNet: a scalable and accurate geometric deep learning potential for molecular dynamics simulation. arXiv preprint arXiv:2210.16518, 2022.
[3] Wang T, He X, Li M, et al. AIMD-Chig: Exploring the conformational space of a 166-atom protein Chignolin with ab initio molecular dynamics. Sci Data 10, 549 (2023).